

研究テーマ2.
赤外ポンプ-プローブ分光法による水素結合錯体の振動ダイナミクス
超高速赤外分光による溶液中の水素結合錯体の振動ダイナミクス
伴野元洋・太田薫・山口小百合・平井聡里・富永圭介
[分子シミュレーション研究会会誌“アンサンブル”Vol. 11, No. 2, April 2009 (通巻46号)から改編]
1. はじめに
水素結合のエネルギーは,一般的な分子間力であるファンデルワールス力よりも大きく,共有結合エネルギーよりも小さい.水素結合による「中間的」な分子間相互作用は,自然界の化学反応において重要な役割を果たす.また,水素結合はフェムト-ピコ秒の時間スケールで解離・生成を繰り返しており,これによる分子環境の揺らぎも化学反応に大きな影響をおよぼす.
水素結合ダイナミクスを観測する手法として,超短パルス赤外(IR)光を用いた時間分解分光は非常に有用である.振動励起された分子は,分子内・分子間エネルギー移動によって励起エネルギーを失い基底状態へと緩和する(振動エネルギー緩和; VER).したがって,VER過程は分子環境を鋭敏に反映する.超短赤外パルスを用いた分光法によって,溶液中[1-6],気相中[7,8]での分子間水素結合錯体のVER過程は研究されている.また,水素結合とVER過程との関係は理論的側面からも研究されている[8].
本稿では,サブピコ秒IRポンプ・プローブ分光を用いて観測した分子間水素結合系の振動ダイナミクスに関して議論する.まず,アルコール中の9-フルオレノン(FL),酢酸メチル(MA)および重水中の酢酸(AA)のカルボニルCO伸縮振動のVER過程を観測した[9-11].AAはOH基が水素結合供与基となりうるが,CO伸縮振動ダイナミクスを観測することでCO基周辺の環境のみを議論することができる.また,フェノール(PhOH)と安息香酸(BA)のOH伸縮振動のVER過程を観測した[12,13].CO基とOH基は分子間水素結合錯体中においてそれぞれ水素結合受容基と供与基となる.環境の異なる振動子に関するVER過程を観測することで,分子間水素結合錯体中の振動ダイナミクスを包括的に議論することを目的とした.
2. 実験
本研究では,時間分解IRポンプ・プローブ分光装置を用いてVER過程を観測した.IR光パルスはチタンサファイア再生増幅器からの出力を光パラメトリック増幅器と差周波発生によって波長変換することで発生させた.IR光は対象とした振動モードと共鳴する波数に調整し,ポンプ光・プローブ光の両方として用いた.プローブ光は分光器によって分散し,MCT検出器で観測した.装置に関する詳細は文献[11]を参照されたい.
3. 結果と考察
3.1. 定常赤外吸収スペクトル
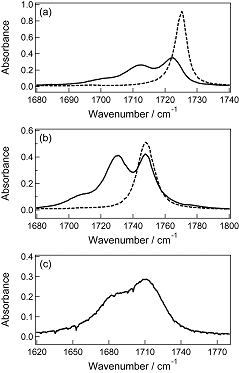 |
| 図1. (a) シクロヘキサン(点線),1-オクタノール(実線)中のFL,(b)四塩化炭素(点線),メタノール(実線)中のMA,(c)重水中のAAの定常IR吸収スペクトル. |
溶液中のFL,MA,AAの定常IR吸収スペクトルを図1に示す.無極性溶媒中では,FL,MA共にCO伸縮振動に由来する単一のバンドが観測されるのに対し,アルコール中のFL, MAでは三つ,D2O中のAAは二つの成分を含んでいた.この結果は,無極性溶媒中ではカルボニル化合物が水素結合を形成していない単量体のみが存在するのに対し,プロトン性溶媒中では複数種の溶質・溶媒水素結合錯体が存在することを示している.量子化学計算を用いた基準振動数計算の結果,アルコール中のFLとMAは,CO基が二つのアルコール分子と水素結合を形成した錯体(1:2錯体),一つのアルコール分子と形成した錯体(1:1錯体),および単量体として存在することがわかった.一方,AAは重水中で1:1, 1:2錯体を形成し,単量体は観測されないと結論づけた.
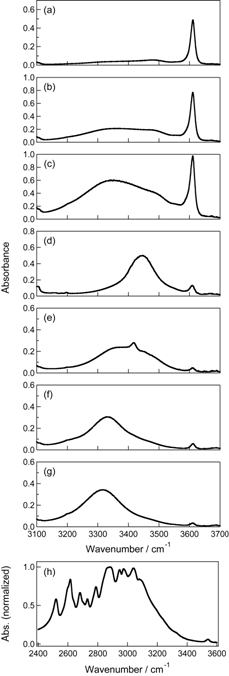 |
| 図2. 四塩化炭素中の定常IR吸収スペクトル.(a) PhOH,0.15 M,(b) 同0.3 M,(c) 同0.5 M,(d) PhOH-ベンゾニトリル,(e) PhOH-アセトン,(f) PhOH-ジエチルエーテル,(g) PhOH-THF.(h) BA-d5 |
続いて,OH伸縮振動に由来する定常IR吸収バンドについて議論する.図2(a)-(c)に四塩化炭素中のPhOHの定常吸収スペクトルの濃度依存性を示す.もっとも濃度が低いとき,3600 cm-1の幅の狭いバンドの相対強度がもっとも大きく,濃度が増すにつれてこのバンドの相対強度が減少し,同時に低波数側の幅広いバンドの相対強度が増加する.同様の結果は四塩化炭素中のメタノールでも観測されており[14,15],この結果から類推して,3600 cm-1のバンドはPhOH単量体に,低波数側のブロードなバンドは複数のPhOHが水素結合で鎖状に連なったオリゴマーに帰属した.図2(d)-(g)に四塩化炭素にPhOHとベンゾニトリル,アセトン,ジエチルエーテル,テトラヒドロフラン(THF)を同時に溶解した溶液のスペクトルをそれぞれ示す.PhOHは,これらの極性分子と水素結合へテロ錯体を形成する.図2(d)-(g)で見られるように,極性分子の種類により,PhOHと極性分子との水素結合強度を反映してピーク位置が異なる.ピークが低波数にあるものほど強い水素結合を形成していると考えられる.
図2(h)に四塩化炭素中のBA-d5のスペクトルを示す.無極性溶媒中でほとんどのBAは環状二量体を形成する[16].OH伸縮振動に由来するバンドは2400-3400 cm-1の波数領域にわたる非常に幅広のバンドと複雑な構造を呈し,バンド形は四塩化炭素,クロロホルム-d,ベンゼン-d6と溶媒を変化させてもほとんど依存しない.このバンド構造はOH伸縮振動と低振動モードとの結合,他の振動倍音・結合音とのフェルミ共鳴,Davydov結合の寄与を含むことが理論計算によって示されている[6,16-18].
以上のように,定常OH伸縮振動バンドは分子間水素結合の強度を鋭敏に反映する.
3.2 時間分解赤外ポンプ・プローブ信号
まず,VER速度を定式化する.フェルミ黄金律によると,分子が始状態iから終状態fに遷移する速度kf←iは,
![]()
と表現される.ここで,分子の状態変化に連動して熱浴の状態はaからa’に遷移するものとし,(HSB)ia, fa’は分子環境の揺動(ハミルトニアンHSBで表示)による,始状態iから終状態fへの遷移を表す項,Ei, Efは状態i, fに対応する分子のエネルギー固有値,ea, ea’は熱浴のエネルギー固有値,Qは熱浴の分配関数をそれぞれ表す.この式を物理的に解釈すると,始状態iと終状態fのカップリングが大きく,エネルギー差が小さく,始状態のエネルギー周辺の状態密度(DOS)が大きいほど,遷移する速度kf←iが大きくなるといえる.
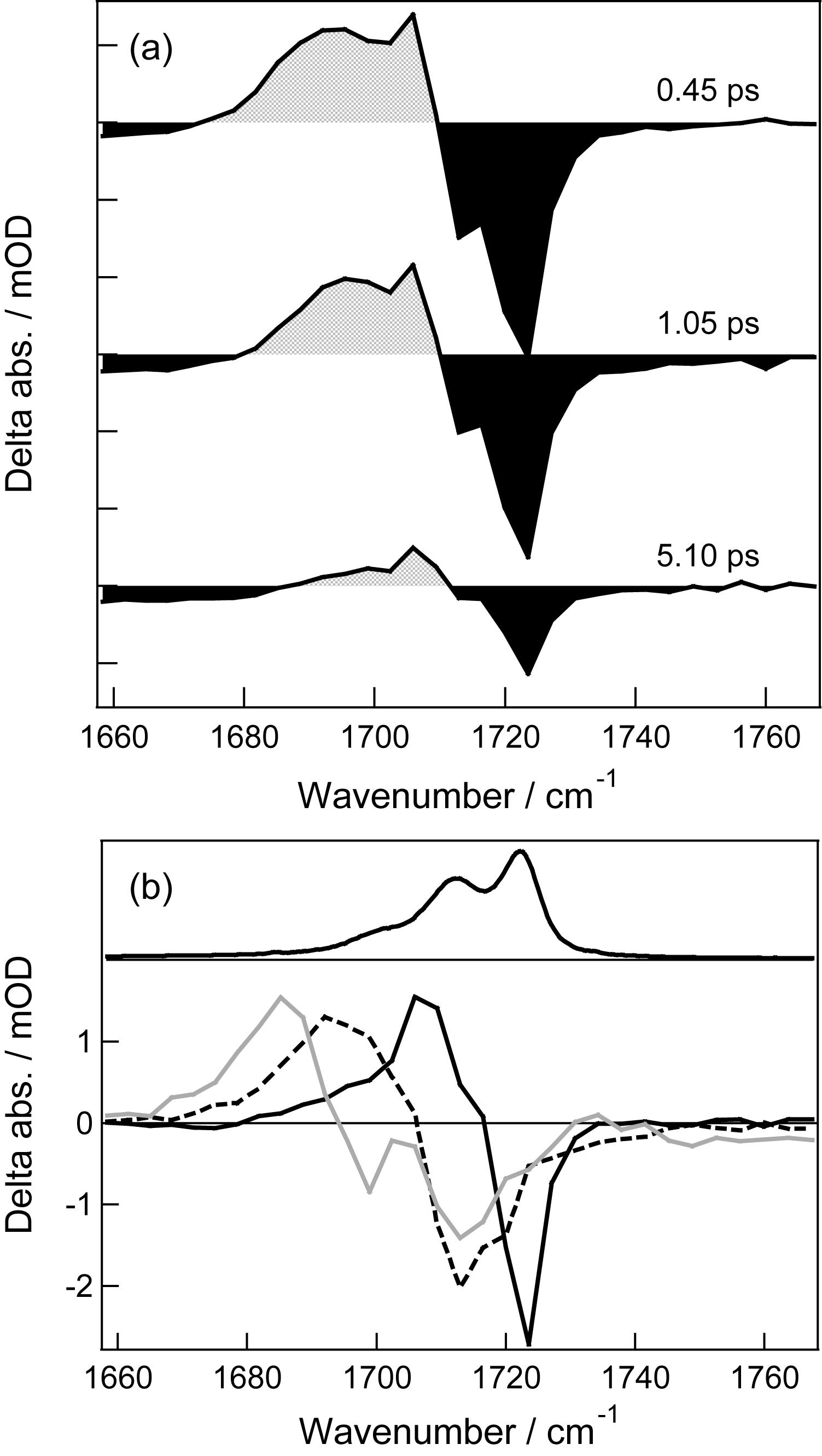 |
| 図3. (a) FL CO伸縮振動励起後の時間分解IR差スペクトル.(b) 時定数0.27 (灰実線),2.3 (点線),4.7 (黒実線) psで減衰する成分のスペクトル.上部は定常IR吸収スペクトル |
1-オクタノール中でFLのCO伸縮振動を励起し,観測されたIR吸収差スペクトルの時間変化を図3(a)に示す.高波数領域に観測された負の信号は定常吸収バンド(v = 1←0)の褪色と励起された分子(v = 1)の誘導放出に,低波数側に観測された正の信号はv = 1状態に励起された分子の遷移(v = 2←1)に対応する.これらの信号の減衰がVER過程(v = 0←1)を表す.得られたポンプ・プローブ信号の遅延時間依存性を解釈するため,三つの指数関数の和をモデル関数としたグローバルフィッティング法を用いて解析した.その結果,三つの時定数は0.27, 2.3, 4.7 psと求められ,それぞれの時定数で減衰する成分のスペクトルは図3(b)のように求められた.2.3,4.7 psで減衰する成分のスペクトル中,負の信号のピーク位置は定常IR吸収スペクトルの二つの高波数側の成分のピーク位置とよく一致する.量子化学計算の結果と合わせて,2.3 psの成分は1:1錯体,4.7 psの成分はFL単量体を励起したことに由来する信号に帰属できた.この結果は,FLが溶媒と水素結合を形成することでCO伸縮振動のVER過程が二倍加速されたことを表す.単量体と1:1錯体で,観測している分子自体は違わないためHSBはほとんど変わらないと考えられる点,CO伸縮振動数が10 cm-1しか違わないため始状態iと終状態fのエネルギー差が大きく違わないと考えられる点から,このVER過程の加速は,分子間水素結合に由来する低振動モードによってDOSが増加したためと考えられる.0.27 psの成分はおそらく1:2錯体に由来していると考えられるが,スペクトル形が定常吸収スペクトルと対応していないため,現時点では明確な議論はできない.
過去のメタノール中のN-メチルアセトアミド(NMA)に関する二次元IR分光の実験で,分子間水素結合の解離・生成は10-15 psの時定数で進行することが観測された[19].この結果から,FL/オクタノール系でも同程度の時間スケールで水素結合の解離・生成が進行すると考えられる.しかし,図3(b)で示したように,本研究での結果では2.3,4.7 psの成分のスペクトルはよく分離されている.したがって,分子間水素結合の解離・生成の進行がVER過程よりも十分に遅いといえる.この結果から,FL/オクタノールの水素結合生成・解離速度はNMA/メタノールと同程度か,それよりも小さいと考えられる.
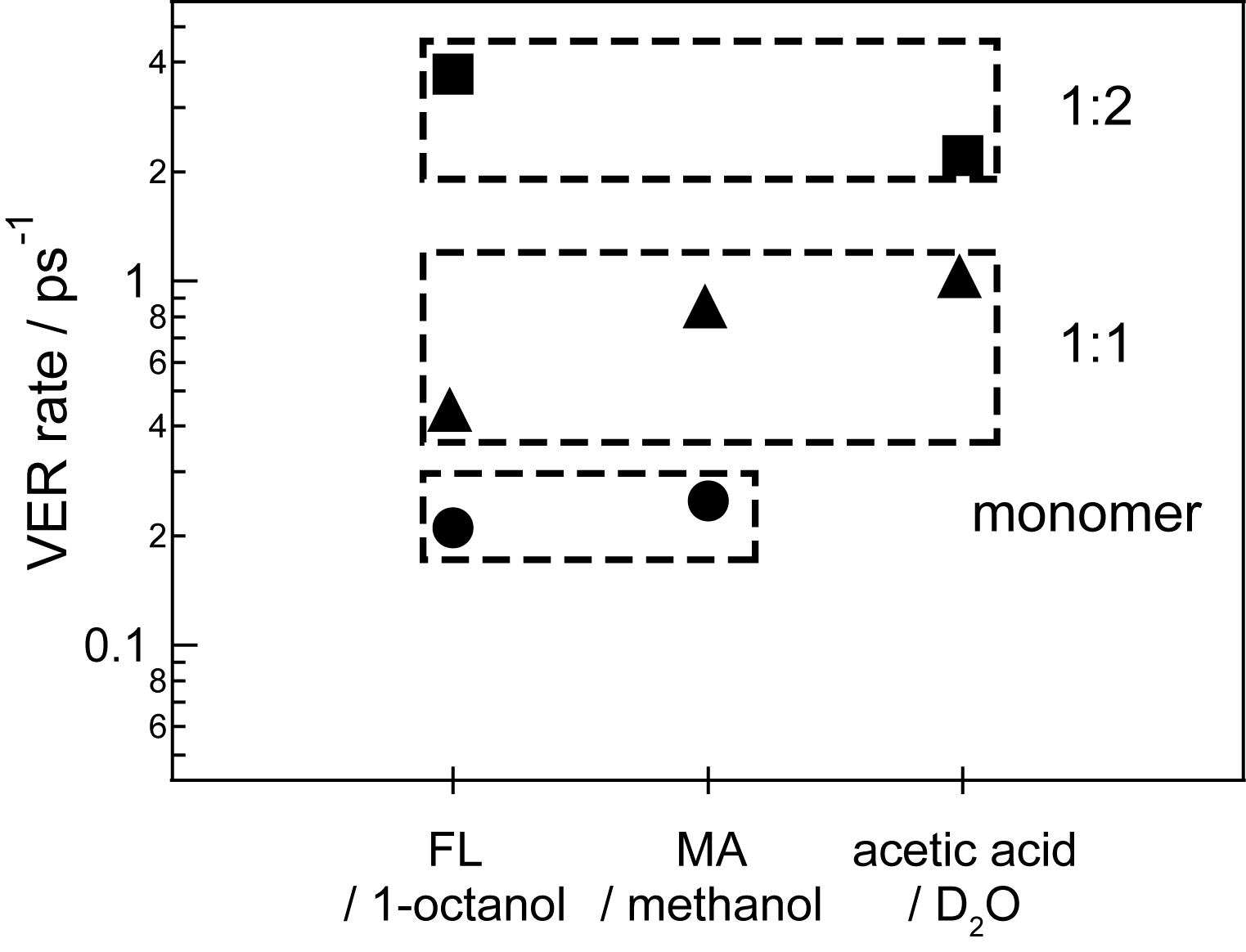 |
| 図4. プロトン性溶媒中でのカルボニル化合物のCO伸縮振動のVER速度定数.単量体(●),1:1錯体(▲),1:2錯体(■) |
FL/オクタノール系と同様に,MA/メタノールおよびAA/D2O系についてもCO伸縮振動励起後のスペクトル変化を観測した.その結果,ポンプ・プローブ信号はMAの場合時定数1.2,4.0 ps,AAの場合時定数450, 980 fsで減衰する成分を含んでおり,これらの成分のスペクトル中の負の信号のピーク位置は,定常吸収スペクトルのピーク位置と一致していた.この結果から,MAで観測された時定数1.2,4.0 psはそれぞれ1:1錯体と単量体の,AAで観測された時定数450, 980 fsはそれぞれ1:2錯体と1:1錯体のVER時間と結論付けた.FLの場合と同様に,MA, AAでも分子間水素結合に由来する低振動モードによってVER過程が加速される.本研究で観測された単量体・1:1錯体・1:2錯体のVER速度を図4にまとめる.
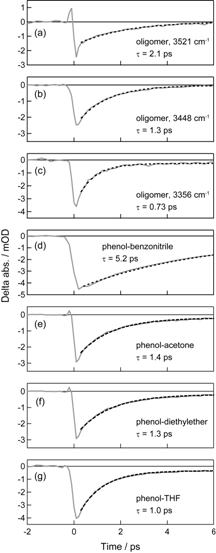 |
| 図5. OH伸縮振動励起後のIRポンプ・プローブ信号.(a)-(c) PhOHオリゴマー.観測波数はそれぞれ3521, 3448, 3356 cm-1.(d)-(f) それぞれPhOHとベンゾニトリル,アセトン,ジエチルエーテル,THFとのヘテロ錯体.時定数は図中に表示. |
PhOHオリゴマーのOH伸縮振動のIRポンプ・プローブ信号の遅延時間依存性を図5(a)-(c)に示す.観測波数が低波数側に移動するにつれてポンプ・プローブ信号の減衰の時定数tが小さくなる.オリゴマー中の水素結合が強くなるほどOH伸縮振動数が減少することを考えると,この実験結果は水素結合が強いオリゴマーほどOH伸縮のVER過程が速く進行することを示しているといえる.また,PhOHと他の極性分子とのヘテロ錯体について,OH伸縮振動のIRポンプ・プローブ信号を図5(d)-(g)に示す.オリゴマーの場合と異なり,ヘテロ錯体のポンプ・プローブ信号のtは,観測した全波数領域において誤差の範囲で一致し,定常吸収バンドのピーク位置が低波数側にあるものほど小さい.この結果も,オリゴマーの場合と同様に,PhOHと極性分子間の水素結合が強い錯体ほどVER過程が速く進行することを示していると考えられる.このように,OH伸縮のVER過程は水素結合強度を鋭敏に反映する.
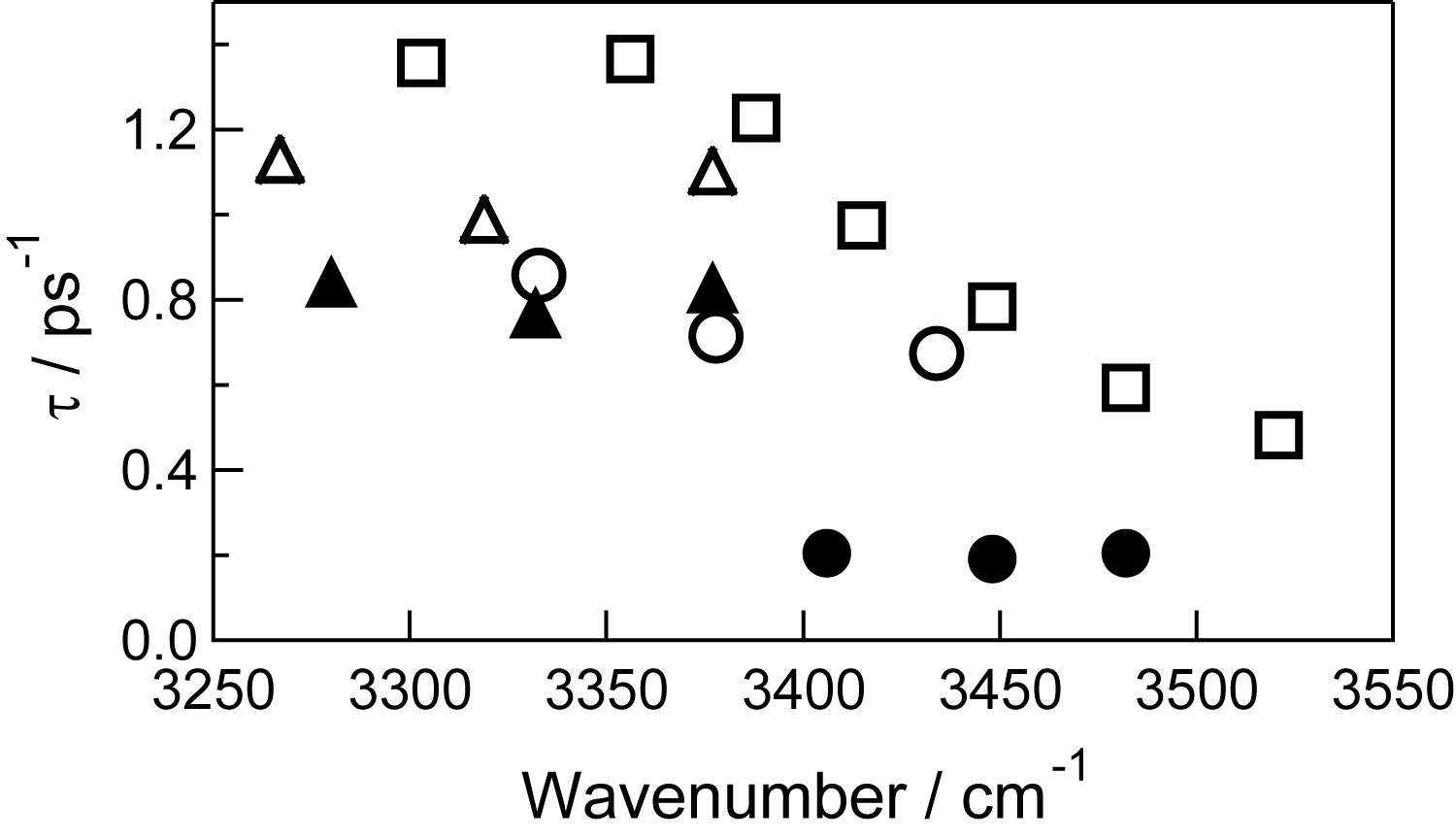 |
| 図6. PhOHのOH伸縮振動励起後のポンプ・プローブ信号減衰の時定数tと観測波数の関係.試料はオリゴマー(□),および,ベンゾニトリル(●),アセトン(○),ジエチルエーテル(▲),THF(△)とのヘテロ錯体 |
本研究で観測したPhOHオリゴマーおよびPhOH-極性分子へテロ錯体のOH伸縮振動励起後のポンプ・プローブ信号は単一指数関数的に減衰する.観測波数と減衰の時定数tとの関係を図6にまとめる.tはオリゴマーでは観測波数に依存し,ヘテロ錯体では依存しなかった.オリゴマーで観測された波数依存性は,振動エネルギー移動後の分子状態fが最初のOH振動励起状態iよりも低エネルギー側に位置しており,OH伸縮振動の波数が小さい分子ほどエネルギー受容状態とのエネルギー差が小さくなるためVER過程が速くなると考えることで解釈できる.あるいは,オリゴマー中での環境の異なるPhOH分子が存在することに由来する可能性が考えられる.オリゴマー由来の幅広い吸収バンドには,OH基が水素結合の受容体かつ供与体となる分子(d),供与体のみとなる分子(g)という二種類の分子種の寄与が含まれている[14,15].これらの分子種ではVER時間が異なると予想されるので,これらの分子種からの寄与が観測波数によって異なると考えることで,tが観測波数に依存するという実験結果を説明できる.一方,ヘテロ錯体でtの波数依存性が観測されなかった結果は,OH伸縮振動の振動数が非常にすみやかに吸収バンド内にスペクトル拡散するためと考えることで説明される.しかし,過去に報告された四塩化炭素中でのフェノール-d・アセトン錯体中のOD伸縮振動に関するtは観測波数に依存しており,本研究でのPhOH・アセトン錯体のOH伸縮振動と異なった挙動を示している[5].このようなtの波数依存性の違いは,溶液中のOH/OD基の振動ダイナミクスを反映しており,非常に興味深い問題である.
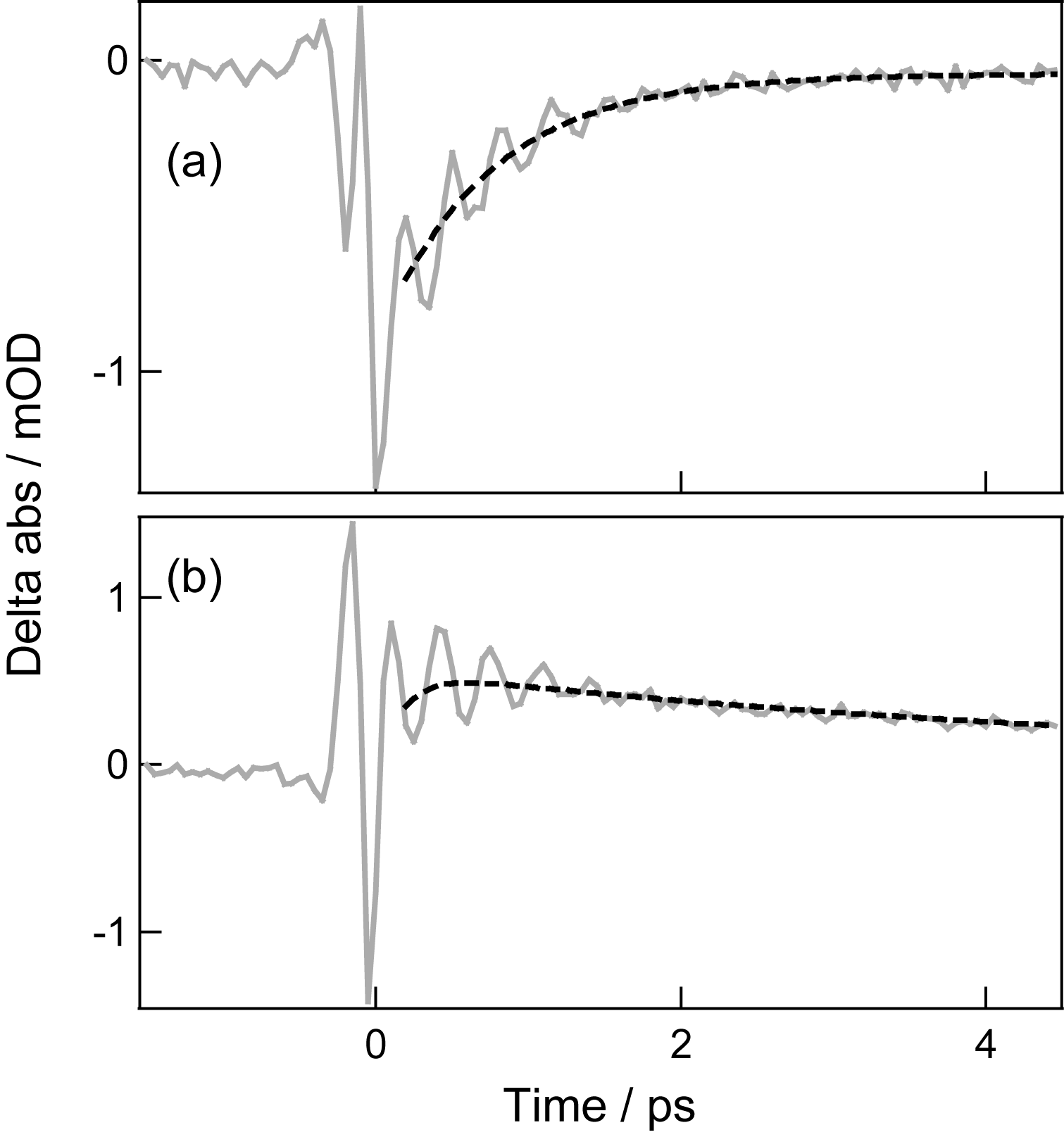 |
| 図7. BA環状二量体のOH伸縮振動励起後のポンプ・プローブ信号の遅延時間依存性.観測波数は(a) 3000 cm-1,(b) 3124 cm-1.点線は指数関数の和によるフィット結果 |
BA環状二量体・OH伸縮振動励起後のIRポンプ・プローブ信号を図7に示す.定常吸収バンドの低波数側では,振動励起によって生じた負の信号が0.73, 13 psの時定数で単調に減衰するのに対し,高波数側では振動励起直後に観測された負の信号が速やかに正の信号へと変化し,この正の信号が減衰していく様子が観測された.これまで,AA環状二量体のOH伸縮振動がIRポンプ・プローブ法によって研究されている[6].この研究結果から類推すると,OH伸縮振動のVER過程は200 fs以下の非常に速い時定数で進行し,0.7, 13 psのダイナミクスはそれぞれ,BA二量体から周辺の溶媒への振動エネルギー分配過程と,BA二量体周辺からさらに遠方へのエネルギー拡散過程を示すと考えられる.
図7に見られるように,BA二量体のOH伸縮振動励起後のポンプ・プローブ信号には約300 fsの周期で振動するビート成分が重畳している.似た挙動はAA二量体のOH伸縮振動励起後のポンプ・プローブ信号にも観測されており[6],OH伸縮振動と分子間振動との非調和結合に由来すると解釈されている.このビート成分を抽出後,フーリエ変換して得たパワースペクトルは100 cm-1付近にピークを示した.この結果は,3000 cm-1の分子内OH伸縮振動が100 cm-1の分子間振動と強く結合していることを示している.量子化学計算を用いた基準振動数計算の結果,100 cm-1のモードは,分子間伸縮振動(112.9 cm-1)か分子間面内変角振動(106.7 cm-1)に帰属できることがわかった[20,21].さらに,OH伸縮振動との非調和カップリング係数を計算した結果,観測された低振動ビートは分子間伸縮振動に由来すると結論付けることができた[13].
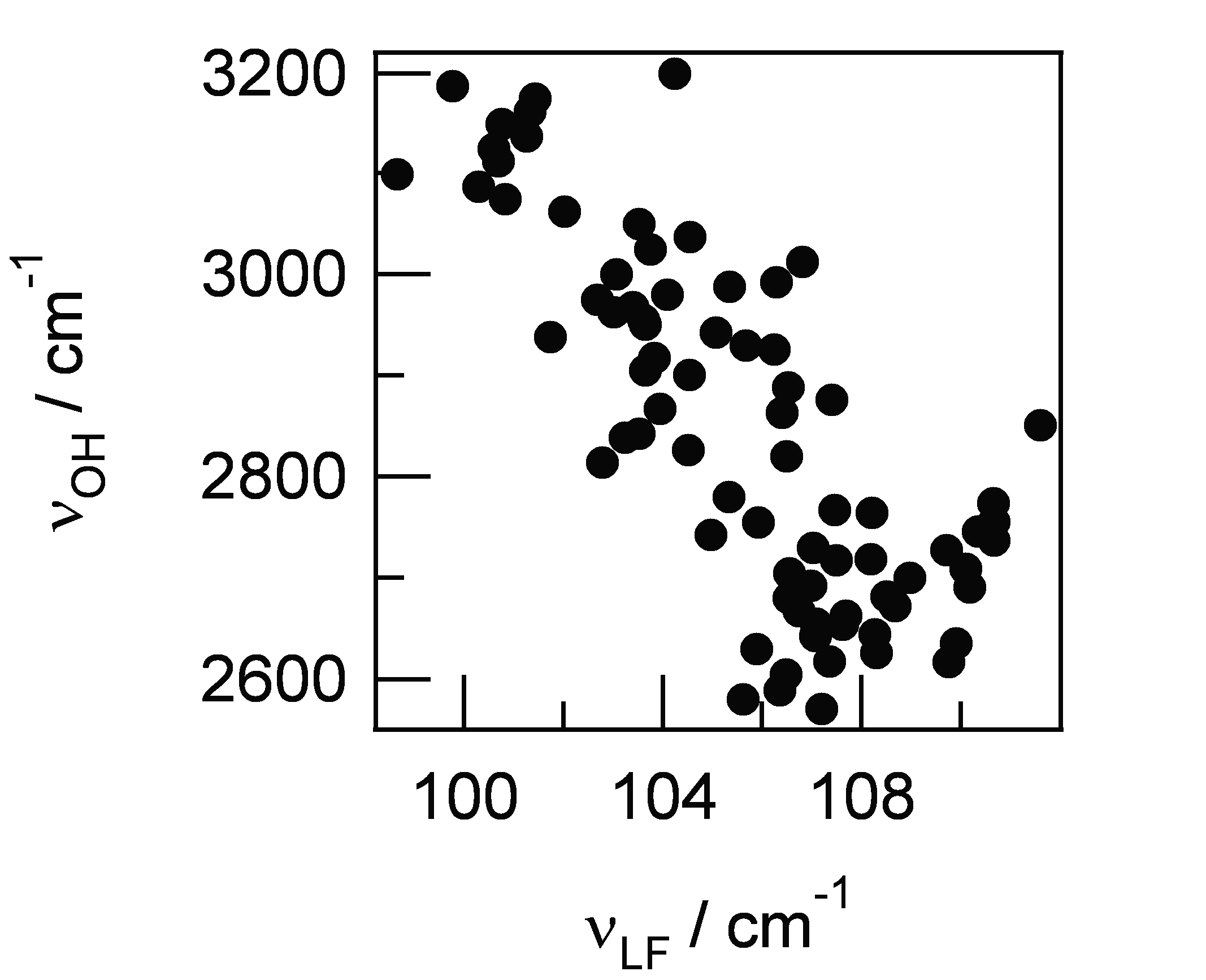 |
| 図8. IRポンプ・プローブ信号の観測波数nOHとビート成分のフーリエスペクトルのピーク位置nLFとの関係 |
分子内OH伸縮振動と分子間伸縮振動の非調和カップリングに関する知見をさらに深めるため,OH伸縮振動の波数と低振動ビート成分の波数との関係を議論する.観測された2600-3200 cm-1のポンプ・プローブ信号の各波数成分からビート信号を抽出し,フーリエ変換することで各波数のビート成分のパワースペクトルを得た.得られたパワースペクトルのピーク波数を,観測したOH伸縮振動の波数に対してプロットした結果を図8に示す. OH伸縮振動の波数が3200 cm-1から2600 cm-1へと低波数方向に移動するにつれて,ビート成分の波数が100 cm-1から108 cm-1へと増加することが見出された.前述したように,分子間水素結合が強くなるにつれ,OH伸縮振動は低波数シフトする.同時に,分子間振動に関しては,振動に関わる水素結合が強くなるため振動数が増加する.この実験結果は,BA二量体の分子内OH伸縮振動の波数と分子間伸縮振動の波数が,水素結合強度に応じて1:1に対応していることを示している.また,分子間水素結合の強さによるBA環状二量体の不均一成分のスペクトル拡散は,1 ps以内の時間領域では完了しないことがわかる.
4.結論
時間分解IRポンプ・プローブ分光法を用いて,水素結合錯体中のCO伸縮振動とOH伸縮振動のダイナミクスを観測した.CO伸縮に関する研究の結果,カルボニル化合物はプロトン性溶媒中で水素結合数の異なる複数の錯体を形成しており,それらのダイナミクスを分離観測することができた.OH伸縮に関する研究結果では,ポンプ・プローブ信号が分子間水素結合の強弱を鋭敏に反映するという知見が得られた.また,BA環状二量体で見られたように,水素結合の強い錯体では分子内OH伸縮振動ダイナミクスの観測が,強く結合した分子間低振動モードの知見を得る上でも有用であることが示された.本研究で得られた知見は,今後分子間水素結合ダイナミクスを研究する上で有効に応用されることが期待される.
参考文献
- W. T. Grubbs, T. P. Dougherty, and E. J. Heilweil, J. Phys. Chem., 99, 10716 (1995).
- S. Woutersen, U. Emmerichs, and H. J. Bakker, Science, 278, 658 (1997).
- N. E. Levinger, P. H. Davis, and M. D. Fayer, J. Chem. Phys., 115, 9352 (2001).
- D. Cringus, S. Yeremenko, M. S. Pshenichnikov, and D. A. Wiersma, J. Phys. Chem.B, 108, 10376 (2004).
- Y. L. A. Rezus, D. Madsen, and H. J. Bakker, J. Chem. Phys., 121, 10599 (2004).
- E. T. J. Nibbering and T. Elsaesser, Chem. Rev., 104, 1887 (2004).
- Y. Yamada, T. Ebata, and N. Mikami, J. Chem. Phys., 121, 11530 (2004).
- S. -I. Ishiuchi, M. Fujii, T. W. Robinson, B. J. Miller, and H. G. Kjaergaard, J. Phys. Chem. A, 110, 7345 (2006).
- S. Hirai, M. Banno, K. Ohta, D. K. Palit, and K. Tominaga, Chem. Phys. Lett., 450, 44 (2007).
- M. Banno, K. Ohta, and K. Tominaga, J. Raman Spectrosc., 39, 1531 (2008).
- M. Banno, K. Ohta, and K. Tominaga, J. Phys. Chem. A, 112, 4170 (2008).
- K. Ohta and K. Tominaga, Chem. Phys., 341, 310 (2007).
- S. Yamaguchi, M. Banno, K. Ohta, K. Tominaga, and T. Hayashi, Chem. Phys. Lett., 462, 238 (2008).
- J. Yarwood, R. Ackroyd, and G. N. Robertson, Chem. Phys., 32, 283 (1978).
- A. Fujii, T. Ebata, and N. Mikami, J. Phys. Chem. A, 106, 10124 (2002).
- P. Novak, D. Vikic-Topic, Z. Meic, S. Sekusak, and A. Sablijic, J. Mol. Struct., 356, 131 (1995).
- D. Chamma and O. Henri-Rousseau, Chem. Phys., 248, 53 (1999).
- K. Heyne, N. Huse, J. Dreyer, E. T. J. Nibbering, T. Elsaesser, and S. Mukamel, J. Chem. Phys., 121, 902 (2004).
- S. Woutersen, Y. Mu, S. Stock, and P. Hamm, Chem. Phys., 266, 137 (2001).
- M. Boczar, K. Szczeponek, M. J. Wojcik, and C. Paluszkiewicz, J. Mol. Struct. 700, 39 (2004).
- J. Antoniy, G. von Helden, G. Meijer, and B. Schmidt, J. Chem. Phys., 123, 014305 (2005).